
Research
Rothamsted Research (2018-2021)
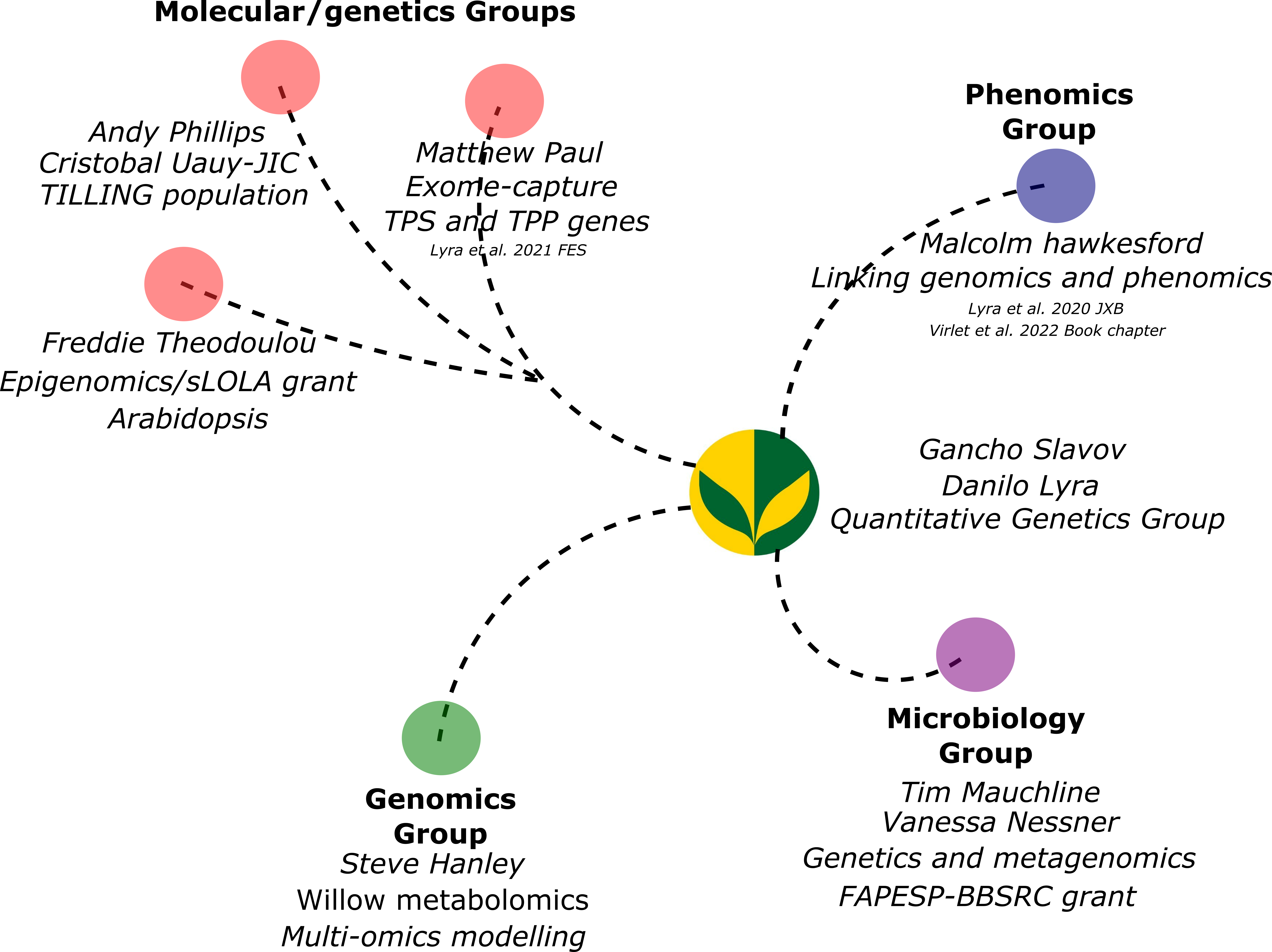
Fig. 1. Quantitative Genetics Network - Rothamsted Research
HOT TOPICS RESEARCH
- Combining high-throughput plant phenotyping and molecular genetics for trait dissection in wheat (link),
- Gene-based mapping of trehalose biosynthetic pathway genes for yield-related traits in wheat (link),
- Development of a quantitative genetics pipeline for the wheat Cadenza TILLING population,
- Integration of genomic and metagenomic data (e.g. rhizosphere microbiomes) for trait dissection in wheat,
- Multi-omics data analysis using artificial intelligence models to study signaling pathways and prediction of phenotypic traits in willow,
- Multi-omic technologies and Big Data analysis to decipher the rules governing the regulation of protein abundance in Arabidopsis (link).

![]()

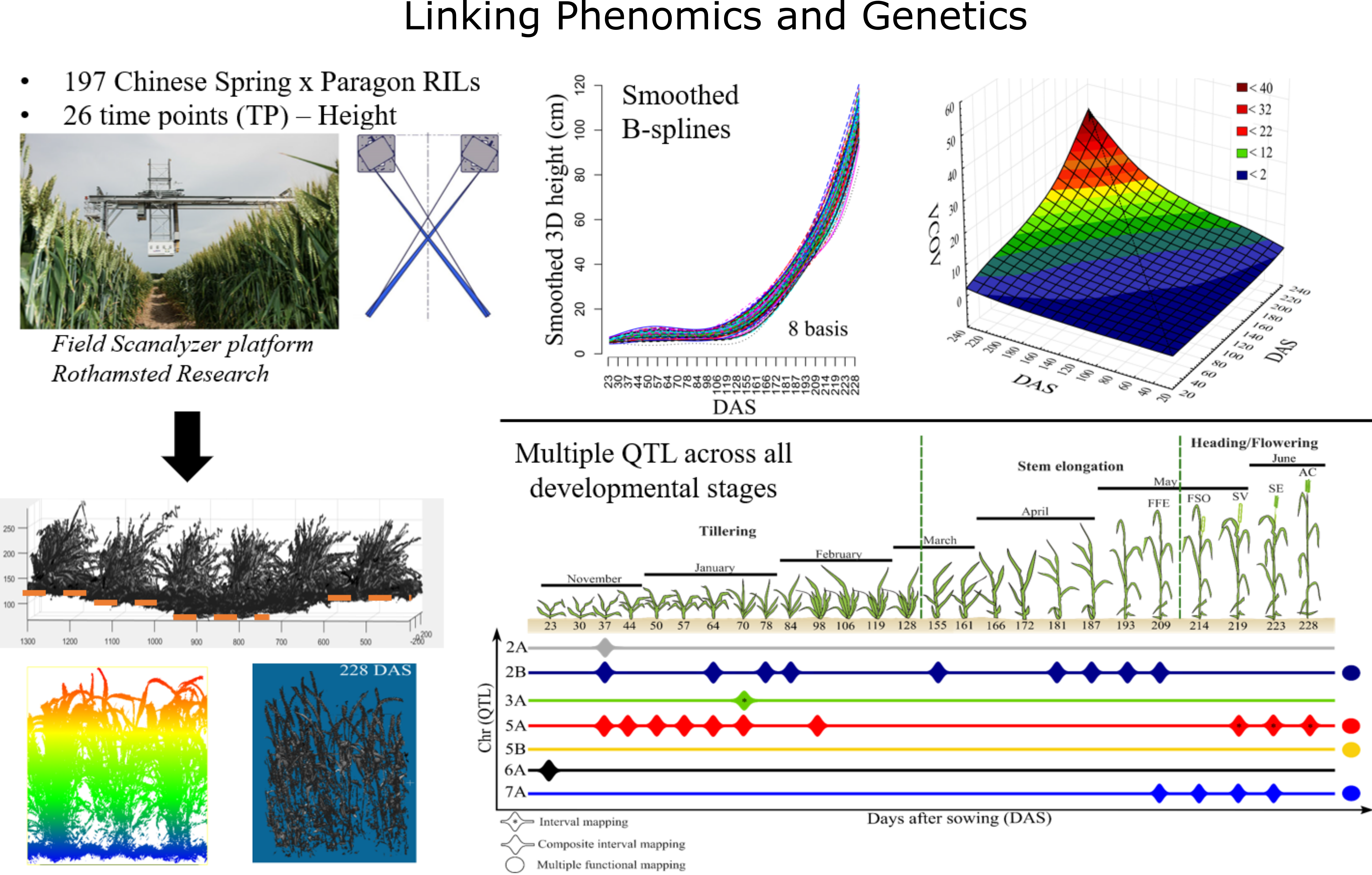
Combining high-throughput plant phenotyping and molecular genetics for trait dissection in wheat
Lyra, D.H. †, Virlet, N., Sadeghi-Tehran, P., Hassall, K.L., Wingen, L.U., Orford, S., Griffiths, S., Hawkesford, M.J., Slavov, G.T. (2020). Functional QTL mapping and genomic prediction of canopy height in wheat measured using a robotic field phenotyping platform. Journal of Experimental Botany, 71(6), 1885-1898.
Abstract Genetic studies increasingly rely on high-throughput phenotyping, but the resulting longitudinal data pose analytical challenges. We used canopy height data from an automated field phenotyping platform to compare several approaches to scanning for quantitative trait loci (QTLs) and performing genomic prediction in a wheat recombinant inbred line mapping population based on up to 26 sampled time points (TPs). We detected four persistent QTLs (i.e. expressed for most of the growing season), with both empirical and simulation analyses demonstrating superior statistical power of detecting such QTLs through functional mapping approaches compared with conventional individual TP analyses. In contrast, even very simple individual TP approaches (e.g. interval mapping) had superior detection power for transient QTLs (i.e. expressed during very short periods). These results will inform the development of an integrated, semi-automated analytical pipeline, which will be more broadly applicable to similar data sets in wheat and other crops.
Digital phenotyping and genotype-to-phenotype (G2P) models to predict complex traits in cereal crops
Virlet, N., Lyra, D.H., Hawkesford, M.J (2022). Digital phenotyping and genotype-to-phenotype (G2P) models to predict complex traits in cereal crops. In: Advances in plant phenotyping for more sustainable crop production (1 Ed.). Editor: Achim Walter (ETH Zürich). Burleigh Dodds Science Publishing.
Abstract Many important agricultural traits, such as grain yield, flowering time, and biomass are quantitative traits. Traditional phenotyping techniques still have been used to collect this data information. However, the revolution of digital phenotyping combined with the new layers of omics and envirotyping tools has been promising to improve selection, accelerating genetic gains. This chapter examines the newest methods used from digital phenotyping tools to predict complex traits in cereals crops. The chapter has two main parts. In the first part, entitled “Digital phenotyping as a tool to support breeding programs”, we will review the secondary phenotypes measured by the high throughput digital phenotyping tools that are potentially useful for breeding. The secondary phenotypes will be considered in terms of the level of integration and complexity regarding their use as single/multi indirect traits for yield prediction, covariate for yield prediction, or integrating over time using growth curve to access new parameters summarising growth events. In the second part, “Implementing complex G2P models in breeding programs”, we will discuss how to integrate data from digital phenotyping into genotype to phenotype (G2P) models to improve the prediction of complex traits using genomic information. We reviewed the current status of statistical models to incorporate secondary traits in univariate and multivariate models as well as how to better handle longitudinal traits.
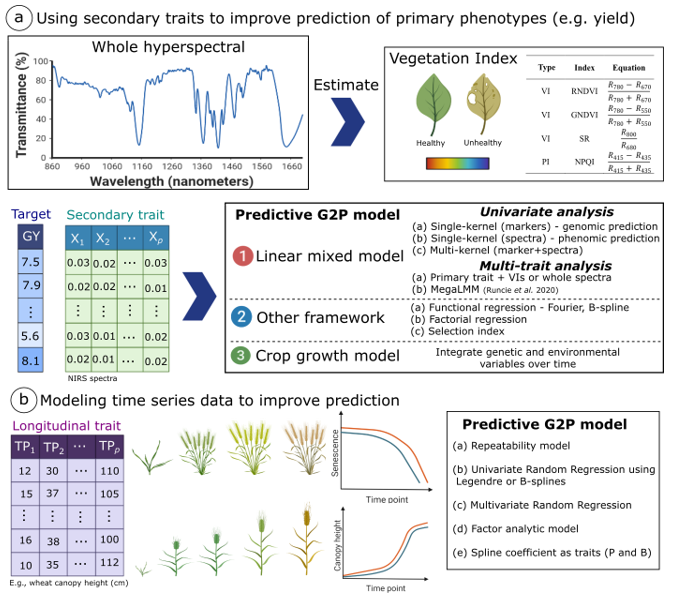
Figure 1. Schematic representation of the integration of HTPP data into genotype-to-phenotype (G2P) predictive models in crop breeding. (a) The first approach is to integrate secondary traits into G2P models to improve the prediction of target or primary phenotypes i.e. grain yield. Secondary phenotypes are represented by whole hyperspectral reflectance data (e.g. near-infrared reflectance spectroscopy, NIRS) and vegetation (VI) and pigmented (PI) index. Secondary traits could be incorporated (1) in a linear mixed models (LMM) using hyperspectral data as predictors (single kernel) in an approach called phenomic prediction or by using multivariate linear mixed model (MvLMM), (2) using other frameworks as functional and factorial regression, and (3) by using crop growth models. (b) The second approach is to explore G2P models for longitudinal traits to capture plant functioning at different stages of plant development. The most common predictive G2P models used for such approach are traditional MvLMM (with specific variance-covariance) and random regression model (RRM).
Gene-based mapping of trehalose biosynthetic pathway genes for yield-related traits in wheat
Lyra, D.H, Griffiths, C.A., Watson, A., Igna, A., Hassani-Pak, K., Molero, G., Reynolds, M., Johnson, R. Hall, A., Paul, M.J. (2021). Gene-based mapping of trehalose biosynthetic pathway genes reveals association with yield-related traits in a spring wheat panel. Food and Energy Security, 10(3), e292.
Abstract Trehalose 6-phosphate (T6P) signalling regulates carbon use and allocation and is a target to improve crop yields. However, the specific contributions of trehalose phosphate synthase (TPS) and trehalose phosphate phosphatase (TPP) genes to source- and sink-related traits remain largely unknown. We used enrichment capture sequencing on TPS and TPP genes to estimate and partition the genetic variation of yield-related traits in a spring wheat breeding panel. Twelve phenotypes were correlated to variation in TPS and TPP genes including plant height and biomass (source), spikelets per spike, spike growth and grain filling traits (sink) which showed indications of both positive and negative gene selection. Individual genes explained proportions of heritability for biomass and grain-related traits. Three TPS1 homologues were particularly significant for trait variation. Gene-based prediction improved predictive ability for grain weight when gene effects were combined with the whole-genome markers. Our study has generated a wealth of information on natural variation of TPS and TPP genes related to yield potential which confirms the role for T6P in resource allocation and in affecting traits such as grain number and size confirming other studies which now opens up the possibility of harnessing natural genetic variation more widely to better understand the contribution of native genes to yield traits for incorporation into breeding programmes.
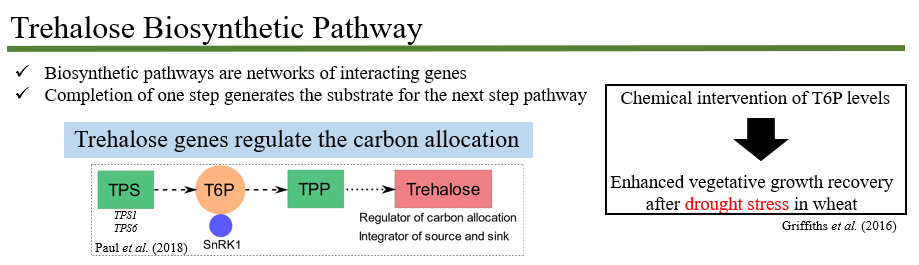
Figure 1. Trehalose Biosynthetic Pathway
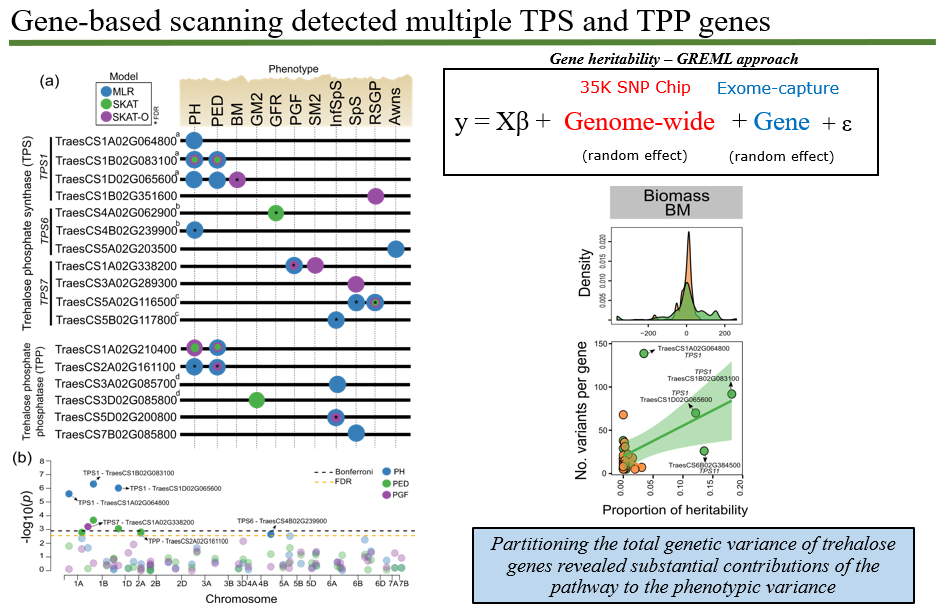
Figure 2. Summary of the gene-based association analysis in the wheat HiBAP panel.
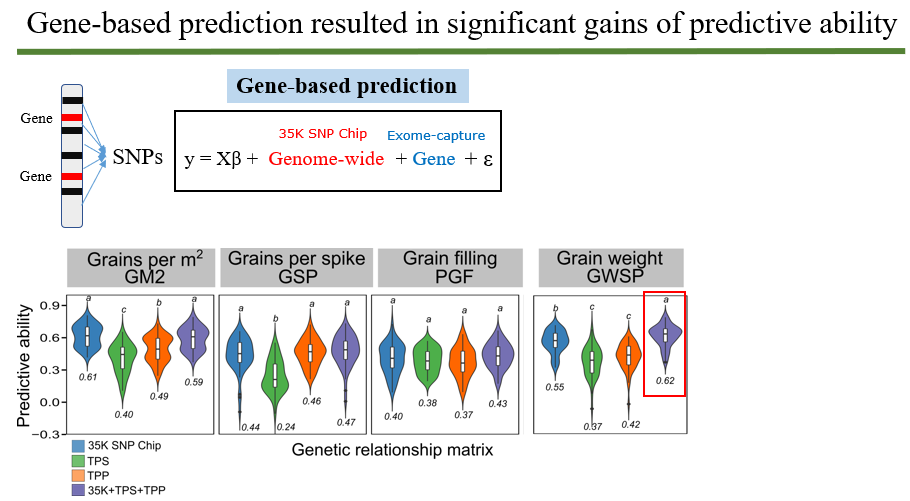
Figure 3. Predictive ability based on genome-wide markers, single gene family and combined effects.
Quantitative genetics pipeline for the wheat Cadenza TILLING population
Lyra, D.H, Virlet, N., Sadeghi-Tehran, P., Thomas, S.G., Huttly, A., Simmonds, J., Uauy, C., Phillips, A., Hawkesford, M.J., Slavov, G.T. The contribution of ultra-rare variants to complex traits in wheat. Unpublished. Poster presented in 6th International Conference of Quantitative Genetics (ICQG6), 2020.
Abstract The relative contribution of rare variants to quantitative genetic variation is largely unclear, despite recent advances in genomics. One reason for this is that natural populations tend to violate the assumptions of genome-wide association and genomic heritability models, with population structure and cryptic relatedness being particularly problematic. To eliminate these limitations, we performed quantitative genomic analyses in an experimental TILLING population of hexaploid wheat (~1100 lines) in which all chemically induced mutations were either rare or unique, starting with a (mostly) uniform genomic background. Using exome re-sequencing data, we partitioned the complete set of ~7M mutations into non-synonymous, synonymous, and non-coding sets, and then conducted association and variance component analyses for three phenotypic traits (i.e. plant height, flowering/heading time, and senescence). By scanning around 77K genes using several analytical approaches developed specifically for rare variants, we identified significant associations for all phenotypes, with most signals coming from non-synonymous and synonymous substitutions. A few well-known genes (e.g. FLOWERING LOCUS T), as well as many others were detected. In addition, up to a third of the variance for all traits could be explained by the cumulative effects of all mutations. We therefore expect that this approach will lead to the identification of candidate genes across a range of phenotypic traits and contribute to the development of novel analytical methodology, ultimately enhancing our understanding of the importance of rare variants in both natural and experimental populations.
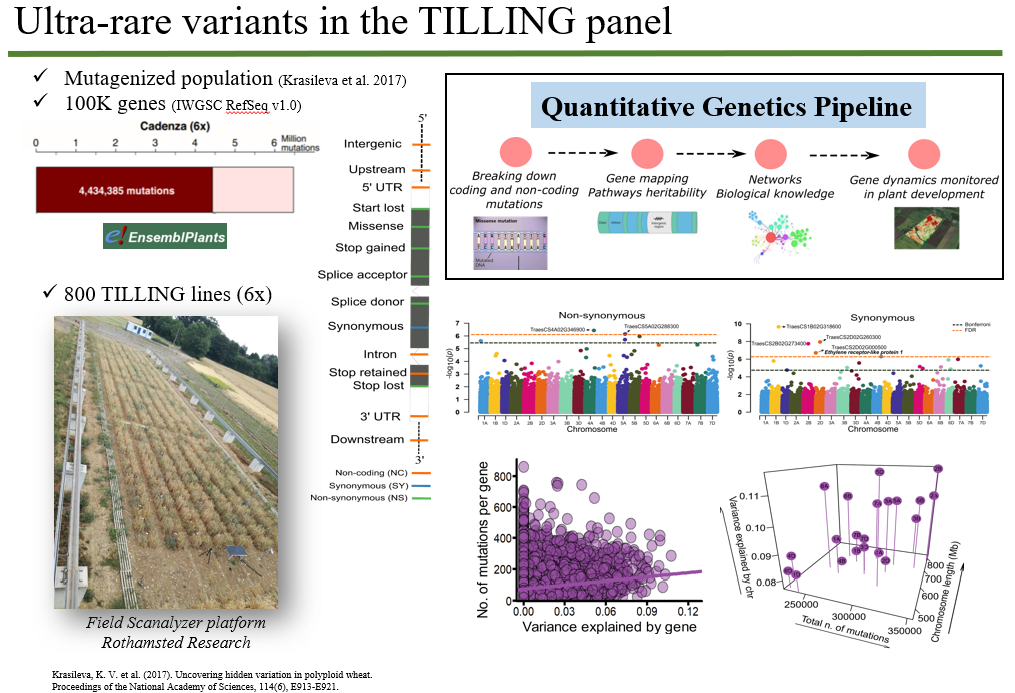
Figure 1. Summary of the quantitative genetics’ analysis of plant height in the wheat TILLING population
Integration of genomic and metagenomic data (e.g. rhizosphere microbiomes) for trait dissection in wheat
Slavov,G., Lyra, D.H, Mauchline, T., Kavamura, V.N. From genomic to metagenomic prediction of complex traits in wheat. Unpublished. Pump-priming Award (UK)-2018.
Abstract Breeding programs have traditionally focused on the aboveground parts of plants. Has modern wheat breeding affected root traits and consequently rhizosphere bacterial communities? Investigating root architectural, morphological and physiological traits among tall and semi dwarf wheat cultivars would increase the understanding of the effect of wheat breeding on rhizosphere bacterial communities.
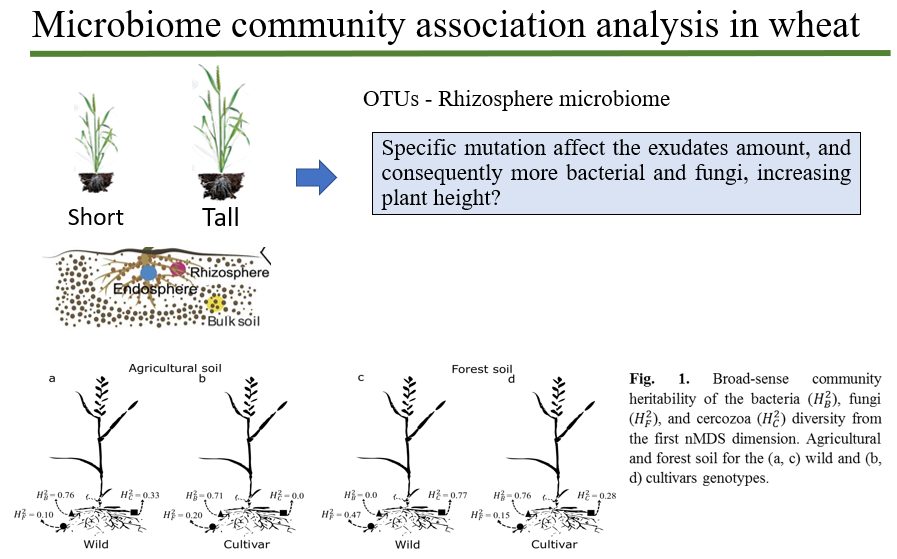
Figure 1. Summary of the rhizosphere communities in wheat
Multi-omic dissection and prediction of adaptive traits in willow (Salix viminalis L.)
Lyra, D.H, Slavov,G., Ward, J, Hanley, S. Metabolome-based association of adaptive traits in Salix viminalis L. Unpublished.
Abstract Willow (Salix viminalis L.) has been used as a bioenergy and biofuel crop across Europe and consists of many bioactive phytochemicals including antioxidants and anticancer. However, the genetic basis of the metabolic diversity has not been yet fully explored, particularly the causal effect of genetic variants on the phenotype acting via the metabolite. We used over sixteen thousand whole-genome variants and a set of candidate gene-derived markers targeting 113 genes to unlock the genetic architecture of hundreds of untargeted metabolites (from three quantification assays) and eight phenology traits in a diverse panel of 220 individuals.
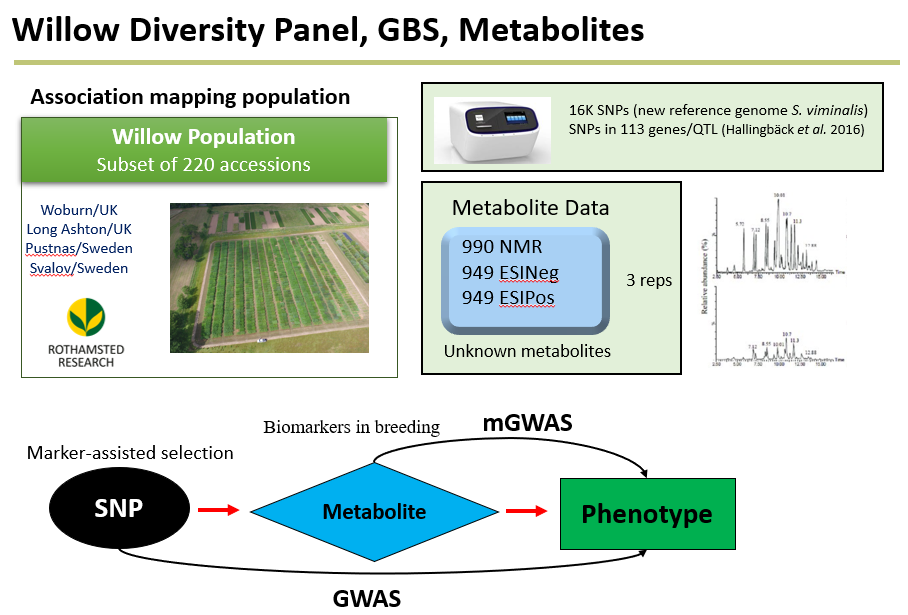
Figure 1. Summary of the metabolome profile in willow